The difference between VIS spectroscopy and NIR spectroscopy
Visible light and infrared
Spectroscopy works for visible light but also in the UV and infrared range. In all cases the same electromagnetic radiation is measured, only the respective wavelengths differ. And yet, the individual ranges are usually used differently: The visible (VIS) range finds its application mostly in color measurement and, thanks to the Fraunhofer lines, also in the detection of various elements. Infrared (IR), on the other hand, is usually used for quantitative analysis, i.e. for determining quantitative ratios in mixtures (e.g. the protein content in beans, or the various fatty acids in vegetable oils). What is the reason for this? Is it simply convention, or are there physical causes?
How do different materials absorb the energy of light?
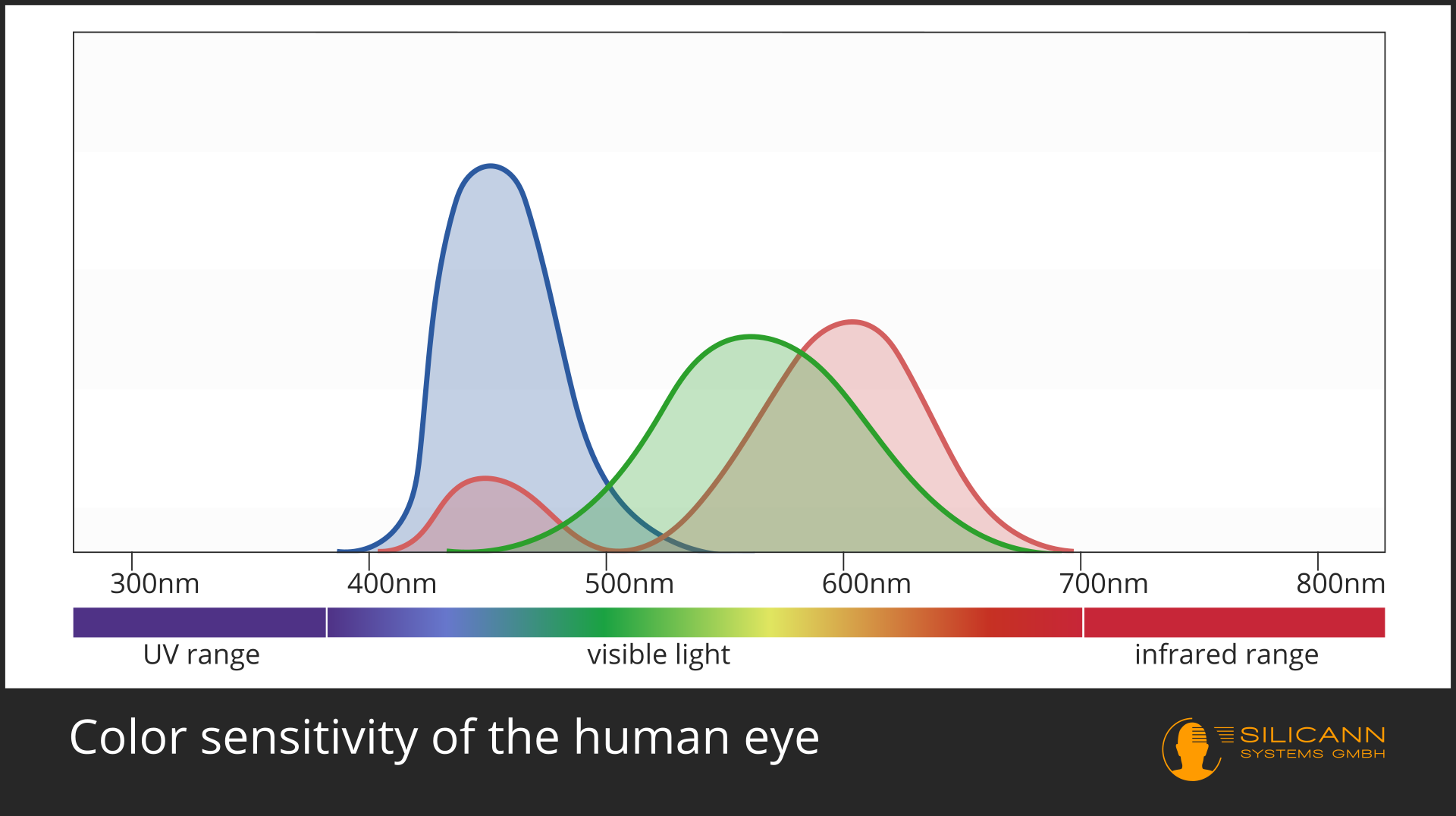
In the case of color measurement the answer is obvious: Of course this is only possible in the VIS range. A spectrometer can analyze the entire wavelengths and calculate exactly the color impression that this spectrum would produce for the human eye. This is not quite trivial, because color does not exist objectively, but is a construction of our visual system: we have three different receptors, each of which has differentl sensitivity to different wavelengths, roughly corresponding to the red, green and blue range. All other colors are then created by interconnections of these receptor signals with each other. Our eyes and those of most other animals have evolved specifically for these wavelengths, because that is precisely the part of electromagnetic radiation in which our star shines most strongly.
Electromagnetic radiation is imagined in the model as a mixture of wavelike and particle radiation. The particles are called photons or quanta. The energy carried by a photon is not always the same, but depends on the wavelength of the radiation.
A general rule applies: The smaller the wavelength, the greater the energy. Visible light has shorter wavelength than infrared, so it is also higher in energy. X-rays and UV radiation have even shorter wavelengths, and are therefore already harmful to the cells of many organisms.
The basis for both phenomena, color as well as UV radiation, which destroys cells, is the fact that atoms can receive or absorb electromagnetic radiation. In doing so, certain atoms only absorb radiation of certain, quite narrow wavelength ranges. This is what gives rise to the Fraunhofer lines, for example.
But why is this like that? Why isn't all incoming radiation absorbed? The answer to this question simultaneously established quantum physics.
The quantum level
Incident photons hit the electrons of an atom, which orbit more or less freely around the atomic nucleus, depending on the element. They absorb the energy of the photons and thereby change their orbit around the nucleus (they change their energy level). According to the classical conception of physics, any arbitrary energy absorption should actually lead to a correspondingly small orbit change. Experiments have shown at the beginning of the 20th century that this was obviously not the case.
At that time, Nils Bohr provided an atomic model that assumed that electrons cannot freely choose their orbits depending on the available energy, but that there are fixed defined orbits between which an electron can only jump completely or not at all. But this also means that it can only absorb very specific amounts of energy, namely always exactly the amount which is exactly sufficient for one (or two or three ...) jumps.
So only the photons whose energy corresponds exactly to the energy needed for the jump of an electron to a higher energy level will be absorbed. The exact magnitude of this energy depends on the material.
We can actually see the effect of these quantum effects with the naked eye: If there were no quantum effects, then every energy level could be reached with radiation. In consequence we would have neither different colors nor the narrow Fraunhofer lines in the spectrum.
IR Spectroscopy
It is similar with molecules. The bonding of atoms to a molecule happens by atoms sharing single quite mobile electrons. These are the valence electrons. Molecules are not tightly bound and immobile, but vibrate around in space in a variety of ways. In a simple model, an atomic bond can be thought of as a metal spring between atoms. The spring holds the atoms together, but allows the bond to stretch and compress. But also, for example, twisting along the bond axis is possible. Depending on the structure of a molecule, there are quite different lines of freedom of the oscillation.
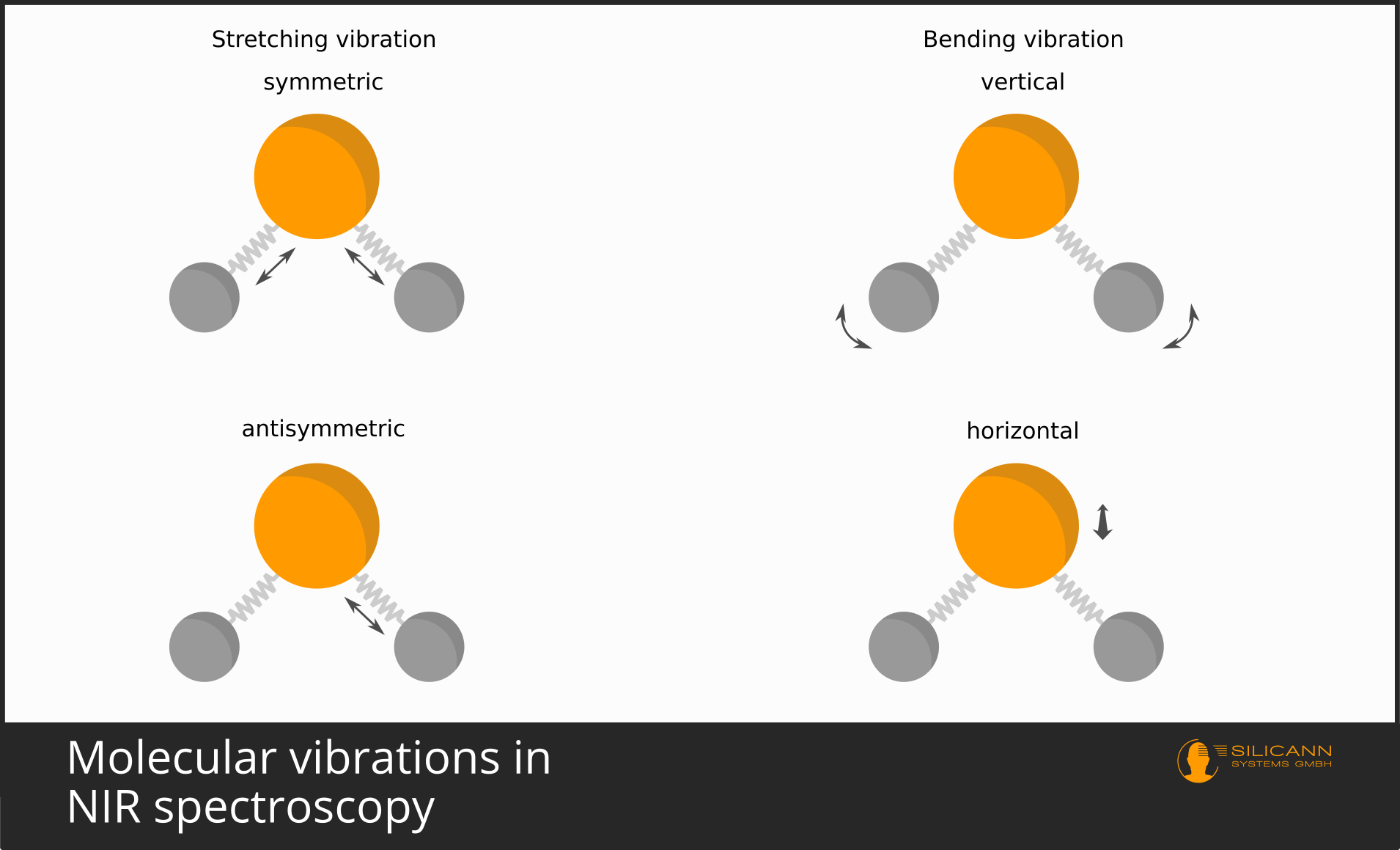
These oscillations, after all, also take place at atomic scales, and therefore the oscillations again are not possible over arbitrary distances, but only in discrete, quantized steps. And, you will guess, again only certain wavelengths correspond exactly to the amounts of energy needed to make the oscillations exactly one (or two, or three) units stronger.
Because there are several directions along which a molecule can vibrate, then very often the individual vibrations overlap and cannot be made out individually in the spectrum. That is why in infrared spectra one usually sees excited bands of vibrations, i.e. somewhat broader wavelength ranges, than the very precise lines in the visible wavelength range.
Fundamental vibration and overtones
It has just been indicated that molecules usually vibrate one energy level more strongly when excited. This jump by one energy level is called fundamental vibration or fundamental oscillation). It occurs most frequently. Accordingly, a sample irradiated with infrared will also cause a large number of molecules to vibrate more strongly in the fundamental mode. Because many molecules have been excited, the fundamental vibration is therefore also almost always clearly visible in the infrared spectrum. For most organic compounds, the fundamental vibration occurs in the mid-infrared region, i.e. at wavelengths between 2500 and 5000 nm.
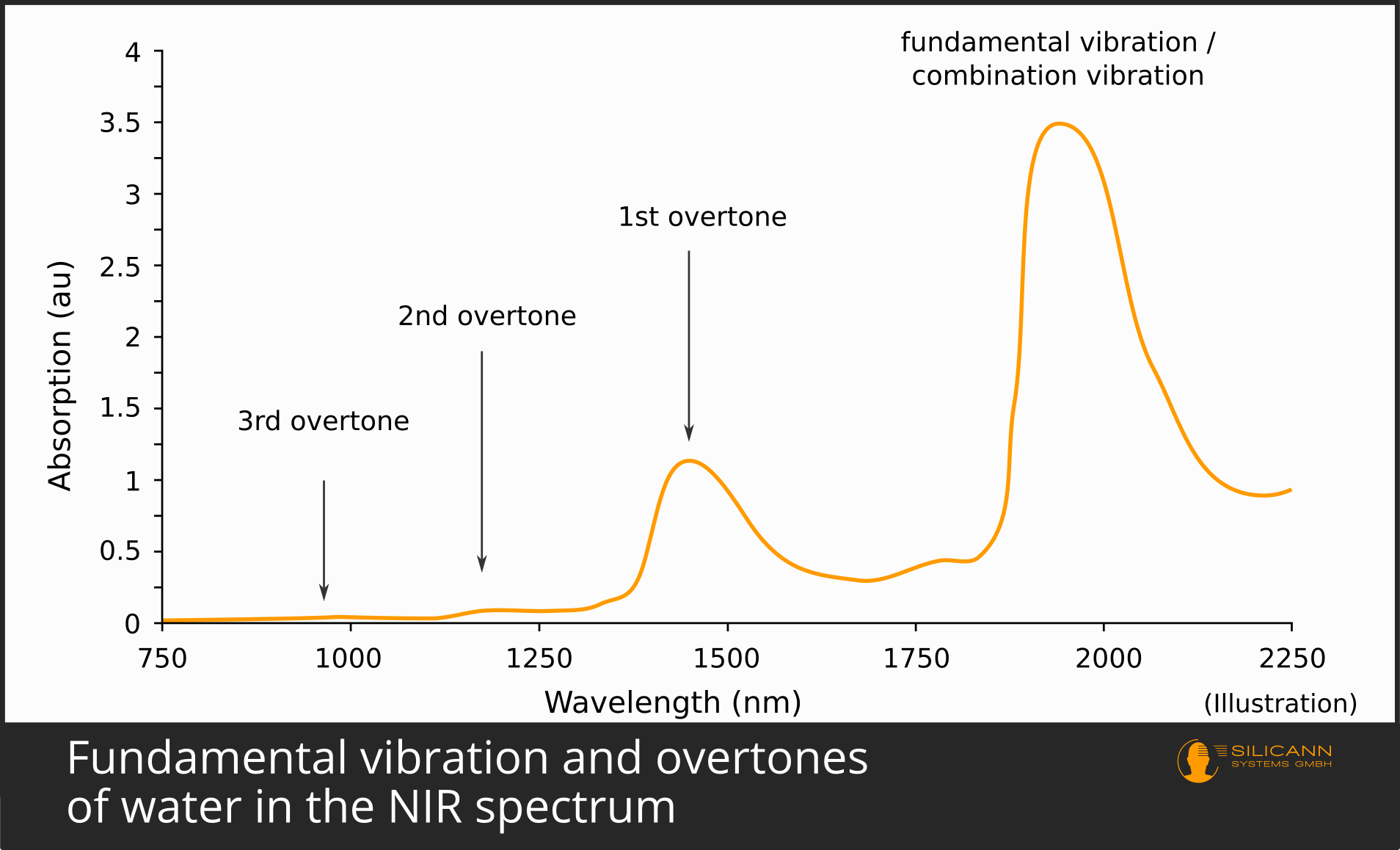
It is much rarer, however, for a molecule to absorb twice the amount of energy at once. This case then becomes visible as the first overtone in the spectrum. The energy again comes from the infrared radiation. Since more energy is required in the case of the overtone, the photons must also supply more energy - they must therefore have shorter wavelengths. Overtones are thus always found at smaller wavelengths relative to the fundamental vibration of the same sample, in the case of organic compounds mostly in the near-infrared range from 800 to 2500 nm.
Because the case of a double jump occurs less frequently, fewer molecules are affected in the sample under the spectrometer. This means that the spike in the spectrum is also significantly smaller than that of the fundamental vibration: Fundamental oscillations are therefore inherently easier to detect than overtones.
The same applies to the second harmonic: it occurs even less frequently, requires even more energy for the jump, the oscillation is correspondingly even shorter wavelength, and in the spectrum it leads to a significantly smaller peak than the first overtone.
Applications of Infrared Spectroscopy
The vibrations of molecules are always characteristic of very specific compounds: The stretching of a CH compound within the molecule would be noticeable as a fundamental vibration at around 3460 nm, a C≡C triple bond at around 4700 nm, etc.
Thus, from the characteristics of a recorded infrared spectrum, conclusions can be drawn about the substance that was measured by the spectrometer, up to and including the precise determination of the chemical structure of an unknown substance. This works particularly well when it is clear that the unknown sample is a single substance.
Very often, especially in industry, infrared spectroscopy is also used for the quantitative determination of contents. A typical example would be the determination of fat and protein content in milk or milk products. In this case, it is a matter of determining the proportions of several mixed substances in a sample.
The procedure in these cases is usually different: A whole series of samples are first examined spectrally and analyzed for their composition, e.g., using classical wet chemical methods, at the same time. If a sufficiently large sample quantity is available, then the correlations between the constituents and the specific spectra can be found out algorithmically. At the end, an evaluation method is then available which can now derive the proportions of fat and protein in the sample purely spectrally.
You might also like
AI-Assisted Development for Spektralwerk Spectrometers
The Spektralwerk API documentation is now available as an MCP server. This facilitates AI-assisted development, particularly in security-conscious companies.


